Chapter One
Uranium is a silver-colored heavy metal in its pure form that is natural, ubiquitous, and radioactive. Small amounts are commonly present in all rock, soil, and materials made from earth's natural substances. Found in air, water, and food, small amounts are consumed and inhaled by all people on the planet every day.
Uranium is produced and used widely in commercial and military applications. U.S. uranium production peaked in 1980 at 21,852 short tons of U3O8 and from that point decreased to 1,534 short tons in 1993 (HHS, 1997b). In 1994, the production again increased, to 1,700 short tons. Uranium is mined from the earth where natural deposits are in concentrations ranging from 0.05 percent to tens of percent by mass. These deposits, called uranium ore, are typically found in sandstone formations. Historically, nearly all U.S. uranium has come from mines in New Mexico, Colorado, Wyoming, Utah, and Arizona.
Uranium is perhaps most recognized in the form of enriched uranium, which is used for nuclear power and nuclear weapons. However, depleted uranium (DU), a by-product of the enrichment process, is used commercially in medicine (radiation shields), aviation (counterweights), space (satellite ballast), and petroleum exploration (drilling equipment). DU has also been used for military purposes. It offers improved defense when used as armor shielding and enhanced power when used in armor penetrating munitions. The United States is not alone in using DU for military purposes. The United Kingdom, Russia, Turkey, Saudi Arabia, Pakistan, Thailand, Israel, and France are developing or already possess weapon systems that contain DU in their inventories (AEPI, 1994).
Two major health concerns are related to the use of DU in military applications: heavy-metal toxicity effects and radiation effects. Heavy metals, such as uranium, lead, tungsten, and others, in sufficient amounts, are toxic to humans and animals. Although much less radioactive than naturally occurring uranium, DU is radioactive and poses a potential health risk from internal and external radiation exposure. DU's toxicity is generally considered to be the greater of the two potential health threats.
For both health issues, many factors will determine whether a health effect may result. Among others, these factors include the toxicological dose (how much and how long), route and magnitude of exposure, and location of embedded fragments. In addition, other factors including age, sex, diet, family history, health status, and lifestyle may affect the overall health consequences of exposure.
Although published health literature dealing directly with DU is scarce, much can be learned from the literature that deals with natural and enriched uranium, which have been studied extensively. This is because both enriched and natural uranium are much more radioactive than DU and, therefore, would have a greater radiation effect than would DU. The chemical toxic effects of natural and enriched uranium are identical to those of DU.
DU is less radioactive than naturally occurring uranium because it has fewer of the more radioactive isotopes per unit weight than does natural uranium. Naturally occurring uranium contains a mixture of three different isotopes:[1] 234U, 235U, and 238U. Uranium-238, 235U, and 234U constitute 99.2745 percent, 0.7200 percent, and 0.0055 percent, respectively, of the weight of naturally occurring uranium.
All of the isotopes of uranium are radioactive and decay into thorium, radium, etc., isotopes of lead (progeny or decay products) until a stable nonradioactive isotope of lead is produced (the complete radioactive decay series for 235U and 238U are shown in Appendixes A and B). This process of radioactive decay also emits ionizing radiation (alpha particles, beta particles, or gamma rays) with each nuclear transformation. U-238, which constitutes more than 99 percent of the mass of natural uranium, is the least radioactive per unit weight. In contrast, 235U exhibits approximately seven times and 234U approximately 18,000 times the radioactivity of 238U per unit weight. The three isotopes of naturally occurring uranium--238U, 235U, and 234U--possess half-lives of 4.5 x 109, 7.1 x 108, and 2.5 x 105 years, respectively. The very long half-life of 238U (4.5 billion years), the most abundant isotope, yields a very low decay rate per unit mass of uranium.[2] Because of the high percentage of 238U and its slow decay rate, naturally occurring uranium is, in fact, one of the least radioactive substances among unstable isotopes on the planet. DU can be up to 50 percent less radioactive than naturally occurring uranium depending on the degree of depletion. The material generally used by the U.S. Department of Defense (DoD) is 40 percent less radioactive than natural uranium. The radioactivity of each uranium isotope in 1 �g of either natural or depleted uranium is shown in Table 1.1.
Depleted uranium has a different isotopic mix from natural uranium. This is because DU is the by-product of fuel- and weapons-grade uranium refining. Nuclear power production and nuclear weapons usually require a greater concentration of 235U, ranging from 2 percent to 90 percent 235U by weight, rather than the 0.72 percent of 235U found in nature (see Table 1.1). To achieve this increased concentration of 235U, naturally occurring uranium is subjected to an enrichment process. The enrichment process increases the percentage of 235U in fuel- and weapons-grade uranium, resulting in enriched uranium. Depleted uranium, which is "depleted" of both 235U and 234U (the relatively more radioactive isotopes), is a residual of the enrichment process.
Table 1.1
Radiological Characteristics of Natural and Depleted Uranium
| Isotope | Half-Life (years) | Alpha Particle Energy MeV (percent) | Isotopic (percent) | Activity (mBq/�gU) | Activity Ratio 234U/238U | Activity 235U/238U |
| NATURAL | 1.00 | 0.048 | ||||
| 238U | 4.468 x 109 | 4.147 (23) | 99.2745 | 12.40 | ||
| 4.196 (77) | ||||||
| 234U | 2.450 x 105 | 4.724(28) | 0.0055 | 12.40 | ||
| 4.776 (72) | ||||||
| 235U | 7.037 x 108 | 4.364 (11) | 0.7200 | 0.60 | ||
| 4.395 (55) | ||||||
| Total | 25.40 | |||||
| DEPLETED | 0.18 | 0.013 | ||||
| 238U | 4.468 x 109 | 4.147 (23) | 99.8000 | 12.40 | ||
| 4.196 (77) | ||||||
| 234U | 2.454 x 105 | 4.724 (28) | 0.0010 | 2.26 | ||
| 4.776 (72) | ||||||
| 235U | 7.037 x 108 | 4.364 (11) | 0.2000 | 0.16 | ||
| 4.395 (55) | ||||||
| Total | 14.80 |
SOURCE: Browne et al., 1986.
The Nuclear Regulatory Commission (NRC) defines depleted uranium as uranium in which the percentage of the 235U isotope by weight is less than 0.711 percent (10 CFR 40.4). The military specifications designate that the DU used by DoD contain less than 0.3 percent 235U (AEPI, 1995). In actuality, DoD uses only DU that contains approximately 0.2 percent 235U (AEPI, 1995).
Radioactivity is measured in nuclear transformations (disintegrations) per second per
unit mass (e.g., becquerels per gram (Bq/g), where a becquerel is equal to 1
disintegration per second). DU has a specific activity of 14.8 mBq/�g.[3] As such, DU's radioactivity is 40 percent less than that of
naturally occurring uranium (25.4 mBq/�g) and orders of magnitude less than that of the
enriched uranium used for nuclear power and weapons (![]() 1,750 mBq/�g).
1,750 mBq/�g).
DU is classified as a low-level radioactive material. In comparison, several consumer products contain radioactive material that also emits ionizing radiation, such as present-day smoke detectors. In medicine, radioactive materials and other sources of ionizing radiation are widely used in the diagnosis and treatment of some diseases.
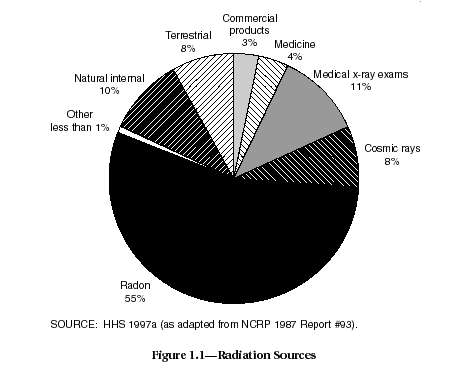
Uranium, in any form, is certainly not alone as a source of ionizing radiation. The world is bathed with low levels of radiation all the time. The sources of radiation dose include radon (55 percent of the total), cosmic rays (8 percent), rock and soil (8 percent), internal exposures from food and water consumed daily in the diet (11 percent), and man-made sources, such as X rays, nuclear medicinal exposure, consumer products, and other sources (HHS, 1997a) (see Figure 1.1).
The hazard of ionizing radiation is derived from the energy it transfers to the matter, including biological matter, through which it travels. This energy is dissipated in the living tissue by diverse molecular interactions, including those with DNA (genetic material) that may result in genetic damage (HHS, 1997a).
All uranium isotopes are primarily alpha particle (a) emitters. These alpha particles will travel only about 30 �m in soft tissue and, therefore, are unable to penetrate paper, glass, or even the dead superficial layer of skin. Consequently, alpha particles present a hazard only if internalized and then only to critical cell structures within the range of the alpha particle from the site of deposition (UNSCEAR, 1982, 1986, 1988). Beta particles have greater ability to penetrate the skin. In most circumstances, beta particles only present a hazard if internalized. In contrast, gamma rays are extremely penetrating. As such, gamma rays present a hazard both internally and externally.
Despite the fact that all uranium isotopes are primarily alpha emitters, other forms of radiation are present. This is because the uranium isotopes decay to other radioactive isotopes (decay products). The natural uranium series is in equilibrium (i.e., the radioactivity of each isotope is the same per unit weight of soil or ores.) When uranium is separated from the ore, the decay chain is broken. Thorium-234 (234Th) and protactinium-234 (234Pa) build up into equilibrium with the 238U within several months. The remaining members of the chain following 234U take thousands of years to reestablish equilibrium and can be considered trivial. Uranium-235 follows the same pattern, and only thorium-231 (231Th) builds into equilibrium rapidly. Therefore, at any given time, some decay products of uranium are present. The decay products are responsible for the presence of beta and gamma forms of ionizing radiation. (See Appendix A for decay schemes of the uranium series.)
Uranium is a heavy metal similar to tungsten, lead, and cadmium. Unlike the radiological characteristics of an element, chemical characteristics of a heavy metal are independent of its isotopic form. All isotopes of uranium exhibit the same chemical behavior (reactivity) and possess identical physical characteristics, such as melting point, boiling point, and volatility. Because naturally occurring uranium, enriched uranium, and depleted uranium vary only in their isotopic mix, they are chemically identical and exert the same chemical effects on the body. Therefore, discussion of the chemical effects of DU will generally refer to "uranium" to cover the natural, enriched, and depleted forms of the metal.
DU possesses certain unique physical properties, such as its remarkable density (19 g/cm3, 1.7 times the density of lead), pyrophoric nature (tendency of fine particles to spontaneously ignite in air), and ductility. Because of these unique characteristics, DU has attracted uses from both the civilian and military communities. To name a few, DU is used commercially in medicine, aviation, space, petroleum exploration, and by the military.
The pyrophoric nature of DU is of special relevance to the health effects resulting from DU's use in munitions and armor. Both the impact of a DU penetrator on a target and the burning of DU produce DU dusts or aerosol particles. In addition to resulting in aerosol particles, when DU burns, the high temperatures created act to oxidize uranium metal to a series of complex oxides, predominantly depleted triuranium octaoxide (U3O8), but also depleted uranium dioxide (UO2), and depleted uranium trioxide (UO3) (AEPI, 1995; CHPPM, 1998). Upon weathering, the nonoxidized small particles and surfaces of remaining uranium metal will also slowly oxidize to those three DU oxides over time (CHPPM, 1998).
Originally it was thought that up to 70 percent of the DU round may be aerosolized upon impact of a DU penetrator on its target or in fires in which DU burns (AEPI, 1995). However, based on more refined testing, the percentage of the original material to aerosolize is now known to range from 10 to 35 percent with a maximum of 70 percent (CHPPM, 1998). The percentage varies according to a number of factors, such as the hardness of the target, velocity and angle of impact, pathway through the target (i.e., what it impacts--engine, DU armor, etc.). If the round easily penetrates a target, as it does non-DU armor, less of it will aerosolize than it does when hitting a nonpenetrable target, such as laminated steel. In many cases of DU hits during the Gulf War, the DU penetrator went completely through (out the other side) the target armor.
Although other industrial compounds exist,[4] the uranium compounds of concern from the military use of DU are primarily limited to the uranium oxides. In the body, the oxides are mostly metabolized to the uranyl ion (UO2++). This discussion is limited to the uranium compounds present in a military environment, namely DU metal and its oxides: U3O8, UO2, and UO3.
The solubility of uranium varies greatly depending on the particular compound and the solvent. Body fluids can dissolve uranium oxides. Once solubilized, uranium may react with biological molecules and exert its toxic effects (Hursh and Spoor, 1973).
In many of its chemical properties, uranium is typical of the heavy metals. Heavy-metal compounds in solution are usually highly reactive and can exert broad cytotoxic effects. Heavy metals, including uranium, exhibit great affinity for biological molecules containing phosphate residues, such as glucose phosphate, phospholipids, and nucleic acids; or sulfhydryl groups, including cysteine, glutathione, and many proteins and oxyanions (oxygen-containing anions). Because of their high affinity for biological molecules, these heavy metals do not exist (except perhaps very transiently) as free ions in biological systems. The heavy metals are present, instead, as complexes with a great variety of molecules (ligands)(Hursh and Spoor, 1973).[5]
For uranium toxicity, the most important oxyanions in biological systems are the carbonate/bicarbonate compounds (Hursh and Spoor, 1973). It has been estimated, for instance, that 47 percent of U(VI) circulating in blood is contained in the inorganic fraction, primarily as [UO2(CO3)2]2 (Durbin, 1984). The compound is stable near neutral pH (the pH of blood) and in this form does not significantly react further with biological molecules. It readily decomposes, however, at more acid pH in urine, with liberation of the reactive uranyl ion.[6]
Once the uranium is solubilized in the blood, the kidney will efficiently excrete about 90 percent of it in urine over approximately three days. Renal excretion of uranium, like that of other heavy metals, is determined by such factors as the filterability of circulating complexes and on the ability of filtered complexes or their decomposition products to be reabsorbed or secreted in the tubule. In one study (Hursh and Spoor, 1973), more than two-thirds of uranium injected intravenously as uranyl nitrate into man was excreted in the urine within one day.[7] Because a major portion of uranium circulating in blood is excreted in urine, increased urinary uranium excretion can provide a sensitive quantitative measure of exposure, especially acute exposure.[8]
It also provides an indication of the amount of exposure over longer periods of time for insoluble uranium compounds, depending on the particular compound and the amount internalized. Embedded fragments present a chronic, steady-state exposure as the metal solubilizes. The background rate of urinary excretion of uranium from natural sources falls into the range of 50-500 ng/day (AEPI, 1995).
The remaining uranium not excreted mostly distributes to bone and soft tissue, including the kidney, liver, lung, fat, muscle, and then, to some extent, to all other organs. In spite of their nonspecific affinity for biological compounds, heavy metals are characterized by specific primary target organs in the body where a change in function is observed. Although uranium in the body distributes to all organs with the main reservoir being the skeleton, the target organ is the kidney, where functional change is observed. The acidification of urine leading to the decomposition of the uranyl-carbonate complexes in tubular urine could help explain the organ-specific reaction of uranium with the kidney (Hursh and Spoor, 1973).
Several different U.S. government agencies regulate and make recommendations about exposure to uranium. Those agencies include the Nuclear Regulatory Commission (NRC), the Department of Energy (DOE), the Environmental Protection Agency (EPA), the Agency for Toxic Substances and Disease Registry (ATSDR), the Centers for Disease Control and Prevention (CDC), and the National Institute for Occupational Safety and Health (NIOSH). The International Commission on Radiological Protection (ICRP), and the National Council on Radiation Protection and Measurements (NCRP) also recommend standards for radiation protection. The EPA, NRC, DOE, and the states promulgate the regulations based on recommendations from the NCRP and ICRP, as well as ATSDR, CDC, NIOSH, WHO, and others.[9]
The organizations that have promulgated basic radiation protection standards for more than 40 years are the NCRP and the ICRP. The dose limit guidelines for human exposure were reduced during this period by about a factor of two as the data on radiation carcinogenesis have developed, mainly from Japanese atomic bomb survivors.
The present guideline for occupational exposure used by NCRP and ICRP is 10 and 20 mSv per year (1 and 2 rem per year) effective dose, respectively. The objective of both organizations is to limit the lifetime radiation dose for maximally exposed persons to 0.7 Sv (70 rem) by NCRP and 1 Sv (100 rem) by ICRP. The NCRP recommends that the lifetime effective dose be limited in 10s of mSv to the value of his or her age (age x 10 mSv, not including medical or natural background exposure). The annual dose should also be limited to 50 mSv (5 rem).
The ICRP recommendation is stated to limit exposure to 100 mSv in five years with no more than 50 mSv in a single year. These lifetime dose figures limit the lifetime fatal cancer risk to less than 3 percent for maximally exposed occupational workers.
Population exposures are also considered because all persons can be exposed in various situations that produce some incremental radiation over what is considered average. Because populations are assumed to have a range of sensitivities, annual exposure is set at a factor of 10 below occupational limits. Thus, a maximum annual effective dose of 5 mSv (500 mrem) is recommended for infrequent exposure, and for continuous or frequent exposure it is recommended that the annual effective dose not exceed 1 mSv (100 mrem). Both these population limits are for exposure to man-made radiation sources other than medical and normal background (NCRP, 1993). The population exposure limits are intended to provide an annual level of carcinogenic risk comparable with other sources, namely one per 10,000 to 1 per 1,000,000. Although this risk is minimal, exposure to radiation, no matter how small, is thought to carry some level of risk.
Military personnel are not classified as radiation workers unless their job specifically qualifies as such. Under battlefield conditions, this classification needs to be determined explicitly so that individuals with duties requiring known exposure to radioactivity, such as cleanup of contaminated equipment, can be monitored and exposure controlled appropriately (NAS, 1997).
In 1944, an urgent need arose for toxicological guidance for the safety of those handling uranium in laboratories and plants for the Manhattan Project. Because this need had to be met before experimental work was available, it was decided to adapt as the maximum permissible level for uranium in air, the value for lead, namely 150 �g per m3. This standard had been used in the lead industry since 1933. This decision was based on the assumption that the radiological toxicity was less important than the chemical toxicity and that lead would be a good surrogate.
Animal experiments with dogs, rats, rabbits, and guinea pigs performed at the University of Rochester are summarized in two reports, one dealing with soluble uranium (Hodge et al., 1949a) and the other with insoluble uranium (Hodge et al., 1949b). This was the most extensive toxicology study of any sort undertaken up to that time. The Rochester experiments were conducted on a scale probably unequaled in the history of toxicology (Hodge et al., 1973). The result was that uranium was classified as a chemical toxin affecting the kidney.
The air concentrations for soluble uranium were based on two established principles (Hodge et al., 1973):
The calculations for human exposure based on these animal studies suggested that the permissible dust exposure concentration for natural uranium in soluble form be set at 50 �g/m3 for an industrial exposure of eight hours per day for a working lifetime. This guideline was used at all of the U.S. government uranium-processing facilities, and extensive replacement of industrial equipment was initiated to comply with this air concentration and to permit high production rates (Christofano and Harris, 1960).
The Rochester recommendation for insoluble uranium compounds in air was calculated on the basis of the then-standard occupational limit for an organ of 15 rem/year (0.15 Sv/yr) and a limit of 100 �g/m3 was proposed. However, the limit of 250 �g/m3 was proposed by the same authors for insoluble compounds based on chemical toxicity (Voegtlin and Hodge, 1953). The authors stated that "the chemical toxicity limit of 250 �g/m3 offered a reasonable margin of safety, and the radiological limit of 100 �g/m3 also offered a reasonable margin of safety." The proposal of two different limits from the same data inevitably caused confusion (Hodge et al., 1973).
The American Conference of Governmental Industrial Hygienists (ACGIH) adopted the maximum permissible concentration of 200 �g/m3 for soluble uranium along with a permissible concentration of 200 �g/m3 for insoluble natural uranium (ACGIH, 1993).
The selection of 3 �g/g of kidney as a de facto permissible standard is based on the radiological limit calculated by ICRP (ICRP, 1959). The ICRP (1959) limit is stated as a whole body content. First the permissible radiation dose to the kidney is calculated and then divided by the fraction of the whole body content in the kidney to obtain the whole body content. The ICRP (1959) value for the whole body content was 0.005 �Ci (185 Bq), and the fraction, at that time assumed to be in the 300 gram of kidney, was 0.065. Thus 0.005/0.33 x 10-6 x 0.065/300 = 3.2 �g/g of kidney.
If this calculation were redone based on today's ICRP annual dose limits of 20 mSv (2 rem) versus the historic limit of 150 mSv (15 rem), and the now well-established fraction of uranium in the kidney, 0.0036 (see Figure 2.5), the permissible body burden would increase by a factor of 2.5.
Current federal and state regulations limit radiation workers' doses to a total effective equivalent dose of 50 mSv/year (5 rem/year) and a committed equivalent dose to any organ of 500 mSv/year (50 rem/year) (10 CFR 20; 10 CFR 835). The EPA has issued regulations for the nuclear fuel cycle that limit the total body dose of members of the public to 15 mrem per year (.15 mSv) and a single organ (except the thyroid) dose to 50 mrem per year (.50 mSv) (40 CFR 190). Currently the NRC cleanup rule specifies 25 mrem per year (.25 mSv) for public exposure.
OSHA, ACGIH, and NRC recommend a limit of 250 �g/m3 for insoluble uranium and 50 �g/m3 for soluble uranium (time-weighted average) for chronic occupational exposure (40-hour work week over the course of a 40-year career) (29 CFR 1910.1000). The short-term exposure limit to natural uranium in the air was set at 600 �g/m3 by the same groups. Based on the Clean Water Act, EPA has proposed a drinking water standard for naturally occurring uranium of 20 �g/L (EPA, 1991). This standard is under review at the EPA (1998).
In the research to be presented, a number of methods have been used to determine the amount and timing of exposure to uranium and DU. In the case of individuals who have been exposed by inhalation or ingestion, researchers use urinalysis. This could be in the form of a 24-hour urine collection or spot urinalysis. (See "Ingestion--Radiological Toxicity," p. 46, and "Diagnosis," p. 58, for a description of the results of urinalysis). Researchers have sometimes used radiological scans including whole body scans to detect exposure, particularly in the case of embedded fragments (See "Embedded Fragments and Wound Contamination," p. 49, and "Diagnosis," p. 58). Additionally, researchers have examined bone ash and tissue samples (usually as part of autopsy). This type of analysis has yielded a great deal of valuable information but obviously cannot be used as a medical tool for diagnosis and treatment of exposed patients. (See "Radiological Considerations," p. 2, "Inhalation--Radiological Toxicity," p. 34, and "Ingestion--Radiological Toxicity," p. 46, for results of this type of research.)
[1]Isotopes of an element have the same number of electrons and protons but have different numbers of neutrons, which give the isotopes different elemental weights.
[2]The radioactive decay rate of any radionuclide is the product of the decay constant, ( = Ln2/half-life in seconds), and the number of atoms, N, present. Decay rate (disintegrations per second) = N.
[3]The symbol m stands for milli (10-3) and � for micro (10-6).
[4]Uranyl salts such as uranyl nitrate, uranyl sulfate, or uranyl acetate and many other compounds of uranium, such as the uranates and halides, are all used in the uranium industry. Some, such as the uranyl salts, are common laboratory reagents. These nonoxide uranium compounds, however, are absent in the military environment (CHPPM, 1998).
[5]This was illustrated, for instance, for inorganic Cd, Hg, or Zn: when injected into the renal artery these metals are instantaneously sequestered by nondiffusible macromolecules, such as plasma proteins, and within fractions of a second become nonfilterable at the glomerulus (Foulkes, 1974). Low-molecular-weight ligands do not significantly contribute to metal binding under the conditions of those experiments, presumably because they are present in plasma in much lower molar concentrations than proteins. The binding of metals by proteins is prevented by higher concentrations of diffusible SH compounds.
[6]Dissolved uranium, similar to other heavy metals, reacts with diffusible chelating compounds, such as EDTA, to form relatively stable and inert complexes in which it can no longer react with other ligands in biological systems. The inert complexes are thereupon filtered and excreted in urine. Such chelating compounds have been therapeutically administered to experimental animals immediately following exposure to uranium. This is called chelation therapy and has proven useful in decreasing tissue levels and increasing excretion of uranium in mice (Domingo, 1990).
[7]The ICRP (1995, p. 220) uses a standard of 80 percent uranium urine excretion in 24 hours.
[8]The measurement of uranium is based on either fluorometric analysis for total uranium mass, radiochemical separation and counting using alpha spectrometry, neutron activation of the 235U with fission track or delayed neutron counting, or inductively coupled plasma mass spectrometry (ICP-MS). The alpha spectrometric analysis permits the isotopic species to be determined in addition to mass. Many data are reported in mass units, typically micrograms, and thus are a measurement of only the 238U, because the mass of uranium is almost exclusively 238U. One �g of 238U = 12.4 mBq = 0.33 pCi.
[9]Standards are presented below for completeness and for benchmarks against which to analyze exposure levels. Standards need to be constantly evaluated in light of new research and information. RAND is neither endorsing nor critically evaluating the standards presented.
Contents
Next Chapter
| First Page | Prev Page | Next Page | Back to
Text |