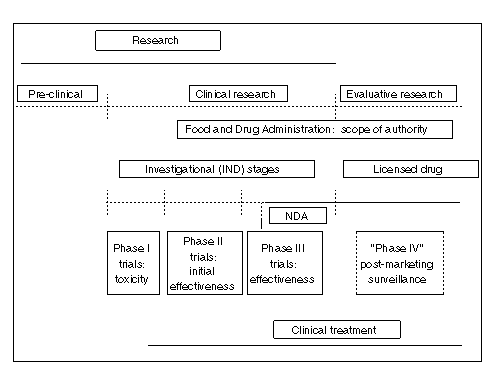
Figure 1--Experiment-Therapy Continuum
The critics of the Interim Rule argue that it was a violation of the basic ethical principles governing research on human subjects in its failure to require informed consent for investigational drugs. This argument is politically salient because of the history of abuse of human subjects in medical experiments, a history shared by both military and civilian agencies of the federal government and by nongovernment institutions and investigators. This history is fresh in our thinking, partly because 1997 was the 50th anniversary of the Nazi doctors' trial and the promulgation of the Nuremberg Code (JAMA, 1996, 276). Also in 1997, President Clinton publicly apologized to the survivors of the Public Health Service's Tuskegee Syphilis Study. These events had been preceded by the report of the Advisory Committee on Human Radiation Experiments (ACHRE, 1996), a 1993 Institute of Medicine report on the health effects of mustard gas and Lewisite (Pechura and Rall, 1993). This history forcefully reminds us of the need for continued vigilance regarding the ethics of human experimentation. But it should neither blind us to learning from that experience nor relieve us from the obligation to examine the particulars of the Gulf War to assess whether and how they differ from these episodes of abuse.
Did the use of investigational drugs under the Interim Rule in the Gulf War constitute research? A definitional excursion is essential to clarify the policy implications associated with the terms "investigational," "research," and medical "practice" or treatment. The Interim Rule applies to "an investigational drug or biologic," and the two waivers granted under it were for drugs FDA classified as investigational. The pertinent questions in this exercise are the following:
The discussion is complicated by two other factors. First, the point of view adopted by the parties to the debate is strongly influenced by their primary referent. These referents include the Nuremberg Code, the Belmont Report, Sec. 1401(c)(1) of the DoD Authorization Act of 1985, and the statutory provisions and implementing regulations of FDCA. Consequently, the discussion sometimes concerns general principles and at other times specific, legally defined applications. At all times, it requires a struggle to disentangle and analyze terms and meanings. Second, it is often the case that substantial subjectivity, even partisanship, characterizes the definitional discussion, making clear that the task is not simply a scientific exercise but is often about conflicts in underlying values.
The continuum is conceptually applicable to all medical innovations, including procedures and therapeutic products. In the development of a new medical procedure, or of a new therapeutic product, such as a drug, biologic, or medical device, a progression can be observed, albeit one involving many iterative steps, from laboratory research to clinical investigation, including clinical trials, and then to clinical practice. This is the medical-scientific dimension of the continuum.
In the case of drugs, vaccines, and medical devices, there is a regulatory overlay. Drugs and biologics are regulated in similar fashion; devices are treated somewhat differently. For drugs, FDA governs the transition from unregulated (preclinical) laboratory research to clinical research involving human subjects by requiring an IND application; it also regulates the transition from the investigational stage to the commercial market by requiring an NDA, which is subjected to extensive FDA review for safety and effectiveness before approval is granted for marketing and which requires postmarketing surveillance after approval. The language of FDCA Sec. 505(a) makes clear the nature of the above controls. It reads:
No person shall introduce or deliver for introduction into interstate commerce any new drug, unless an approval of an application filed pursuant to subsection (b) or (j) is effective with respect to such drug.Finally, the informed consent overlay governs the protection of human subjects: This overlay includes FDA regulations for drugs (21 CFR 50; 21 CFR 56) and the DHHS rule for NIH and CDC clinical research (45 CFR 46); the latter rule, also known as the Common Rule, has been adopted by all federal government agencies.
These three overlays on the experiment-therapy continuum are not described by a clear set of terms having universal acceptance, unambiguous definitions, and consistent applications. Terms that are roughly comparable are not synonymous. Although the two poles of the continuum--the solely experimental and clearly established therapy--are distinct, the territory between them is a proverbial gray zone; no bright orange line divides activities along the continuum.
The continuum is portrayed graphically in Figure 1, which depicts "research" and "treatment" (at the top and bottom of the figure) as overlapping. We can say the following about this depiction. First, research encompasses preclinical, clinical, and evaluative activities occurring after a medical innovation is introduced to clinical practice. But not all clinical research is therapeutic, i.e., intended for the benefit of the individual research subject. Phase I and Phase II trials may or may not benefit the individual human subject, who is a volunteer, but may be intended solely to generate scientific knowledge of benefit to future patients. However, much clinical research is intended to generate both scientific knowledge and therapeutic benefit to the individual subject of research. Moreover, much therapy clearly lies beyond the research stage. For clinical procedures, however, no clear demarcation of the boundary between research and treatment exists, whereas in the case of drugs, the boundary is defined as an artifact of FDA regulations.
Figure 1--Experiment-Therapy Continuum
Second, FDA regulatory authority encompasses all clinical research on therapeutic products under its jurisdiction (drugs, biologics, medical devices)--the "investigational" (or IND) stage; the period after submission of a new drug application (or NDA), during which a drug is evaluated for licensing and introduction to commercial marketing; and postmarketing surveillance (Phase IV studies) after approval. Licensing is based on clinical evidence of safety and effectiveness, but also involves determinations about the adequacy of the manufacturing processes and capabilities, as well as considerations related to labeling.
Third, some research and clinical evaluation continues after a drug or procedure has moved beyond the research stage, has been introduced into clinical practice, and has achieved recognized status as established therapy. This is represented here as "evaluative research" as it applies generally to all medical innovation, the health services research domain of effectiveness and outcomes research, and as "Phase IV" research as it pertains to continuing studies of FDA-approved drugs and vaccines.
This means that the person involved should have legal capacity to give consent; should be so situated as to be able to exercise free power of choice, without the intervention of any element of force, fraud, deceit, duress, over-reaching, or other ulterior form of constraint or coercion; and should have sufficient knowledge and comprehension of the elements of the subject matter involved as to enable him to make an understanding and enlightened decision. This latter element requires that before the acceptance of an affirmative decision by the experimental subject there should be made known to him the nature, duration, and purpose of the experiment; the method and means by which it is to be conducted; all inconveniences and hazards reasonably to be expected; and the effects upon his health or person which may possibly come from his participation in the experiment.
The duty and responsibility for ascertaining the quality of the consent rests upon each individual who initiates, directs or engages in the experiment. It is a personal duty and responsibility which may not be delegated to another with impunity. (JAMA, 1996; p. 1691.)
The commission was charged by the National Research Act of 1974 "to identify the basic ethical principles that should underlie the conduct of biomedical and behavioral research involving human subjects" and to develop guidelines to ensure that these principles were adhered to in such research. The report identified three basic ethical principles--respect for individuals, beneficence, and justice--that should underlie the conduct of biomedical and behavioral research involving human subjects:
The report then articulated three applications of these principles--informed consent, risk-benefit evaluation, and selection of subjects:
For the most part, the term "practice" refers to interventions that are designed solely to enhance the well-being of an individual patient or client and that have a reasonable expectation of success. The purpose of medical or behavioral practice is to provide diagnosis, preventive treatment or therapy to particular individuals. By contrast, the term "research" designates an activity designed to test an hypothesis, permit conclusions to be drawn, and thereby to develop or contribute to generalizable knowledge (expressed, for example in theories, principles, and statements of relationships). Research is usually described in a formal protocol that sets forth an objective and a set of procedures designed to reach that objective.However, the commission did state a general rule that, in situations where research and treatment are carried on together, "if there is any element of research in an activity, that activity should undergo review for the protection of human subjects."
Two appendixes in the report wrestled with the research-treatment distinction. Sabiston addressed the "boundaries between biomedical research involving human subjects and the accepted or routine practice of medicine," with an emphasis on surgery. "As one pursues this subject [of boundaries]," he wrote, "it becomes evident that there is no dividing line which can be consistently agreed upon by any group of authorities on the subject" [emphasis added]. In Sabiston's view, "such an arbitrary division [between medical practice and research] is simply impossible, at least if determined on a rational basis." Although medical practice that was "established beyond reasonable doubt" and research that was "clearly experimental" were easily distinguished, Sabiston found "a definite `gray zone' in which practice and research overlapped and objective classification was extraordinarily difficult."
Robertson addressed the legal implications of physician activities "that occur on the boundary between research and the accepted practice of medicine." He uses "on the boundary" and "boundary activity" synonymously with "innovative therapy," characterizing the latter as activity that subjects patients "under the guise of therapy to risky, untested procedures without the safeguards that apply to experimentation." Although the phrase "on the boundary" implies a clear demarcation between research and accepted practice within the context of protocol-based research, Robertson was actually using this term, and that of innovative therapy, to describe that which is neither protocol-based research nor established therapy, but activity that appears to be "on the edge" of these activities. This usage is reasonable given that the commission was concerned about procedures that departed "in a significant way" from standard practice but that were not conducted according to a research protocol. Such procedures were experimental in that they constituted both a potential and real source of abuse of human subjects. Hence Robertson's discussion is not pertinent to the current discussion.
Parenthetically, it is important to note that no attention was given in the Belmont Report to FDA or its regulations for drug development. The report made no mention of the fact that, under FDCA regulations, radically experimental drugs or vaccines could not be used for research or treatment without a rigorous, FDA-approved protocol. The problems the Belmont Report addressed did not lie in this direction. The report certainly did not address the hypothetical, which became real in the Gulf War, in which a product, such as PB, licensed by FDA as safe and effective for one indication but in a late "investigational" stage for a different indication, was proposed for use as "treatment" (or pretreatment) in military combat. Neither did it address the BT situation, where an IND vaccine routinely used for protective purposes for at-risk individuals was being proposed for similar DoD use. Thus, although the report provides the conceptual framework for bioethicists in their reasoning about the issues under discussion, that framework is not sufficiently developed to embrace these issues from the perspective of drug development and use.
An early ethical defense of the interim rule appeared in the Hastings Center Report. Howe and Martin (1991),[1] briefly reviewed the CW/BW threat posed by Iraq, the "compounds" (referring in general to drugs and vaccines and, in particular, to PB and BT) available for protecting service personnel from these threats, the investigational status of these compounds under FDCA, and their scientific and clinical status.
They then posed two ethical questions:
Should service persons be given these [investigational] compounds? and, if the answer was yes, Should servicepersons be given the opportunity to grant or withhold their consent, as they would if they were research subjects?Extensive review of the data by DoD, FDA, and HHS had resulted in a decision to give the compounds to service members, to inform them of the compounds' investigational status, including side effects and risks, but not to obtain their informed consent.
A major justification put forward by Howe and Martin for this decision was that the use of these compounds was for "preventive or therapeutic treatment as opposed to research." They found support for this distinction in the Belmont Report and the Declaration of Helsinki (World Medical Association, 1997). The former states that treatment usually involves interventions intended solely for the benefit of a particular individual. The fact that "some forms of practice" provide benefits, to others, i.e., knowledge of a general nature, "should not confuse the general distinction between research and practice." The Helsinki Declaration distinguished between medical research whose purpose was "essentially diagnostic or therapeutic for a [given] patient" and that which was intended to be "purely scientific and without direct diagnostic or therapeutic value" to those subjected to it.
Howe and Martin also noted that, although the Nuremberg Code was explicit about the "absolutely essential" requirement of voluntary consent to human experimentation, it was silent regarding clinical research. Using the compounds in question without informed consent, they argued, was consistent with the spirit, if not the letter, of other parts of the Nuremberg Code regarding the conduct of experiments to avoid "unnecessary physical and mental suffering" and to protect against "even remote possibilities of injury, disability, or death."
But since the primary purpose of the use of these compounds in the Gulf War was not research, "the degree, if any, to which their use should be construed as experimental, even according to the two codes that address therapeutic research [Belmont, Helsinki] is open to question." Nevertheless, because these compounds were classified as INDs under the FDCA and "because some generalizable data will be collected concerning the effect of using these compounds," the authors acknowledged "a small research dimension" associated with their use.
The compounds were considered investigational, Howe and Martin argued, for two reasons: first, "they had not been studied to the extent normally required for drugs' commercial use;" and second, because exposing human subjects to the chemical and biological agents that the compounds were intended to protect against would be unethical. However, given that the safety and protective effectiveness of these compounds might differ, "the ethical justification for using each compound before or during combat must be separately determined" (i.e., on a case-by-case basis). It was necessary, therefore, that, in addition to DoD review, experts outside the DoD review both the scientific merits and ethical bases underlying DoD's proposed use of these compounds. Thus, FDA and DHHS conducted external review of these compounds. The conclusion of all parties was that the potential benefits of use were "substantial" and the risks of harm were "extremely small."
The ethical issue then turned on whether service personnel "should have the option of giving or withholding informed consent." The authors then elaborated on military research ethics. No differences between military and civilian research subjects were justified in peacetime. However, "the ethical context shifts," when investigational compounds are used for service personnel in combat or combat-ready situations:
The principle of preventing unnecessary harm to servicepersons (and enhancing the mission by protecting all servicepersons) overrides all other values [emphasis added]; and the principle of respect for persons is fulfilled by maintaining the military's explicit and implicit promise to servicepersons to protect them from unnecessary harm. (Howe and Martin, 1991, p. 22.)Respect for persons, which differs from allowing individuals to grant or withhold consent, is thus predicated on the military keeping its promise to protect individuals. It is also accomplished, insofar as possible, by fully informing service members about the investigational compounds they will be taking:
They [service personnel] should be informed that these [compounds] have not been tested vigorously under battlefield conditions; they should be told of the risks and expected benefits so far as they can be identified; they should be warned that the expected benefits do not include prevention of all harms from chemical or biological attack; and they should be made familiar with known side effects of the compounds. (Howe and Martin, 1991, p. 22.)These shifts from the ethical principles that apply in civilian situations result from the unique obligations of service personnel to their country and to those with whom they serve. Service personnel "are willing to sacrifice their lives if necessary to benefit fellow servicepersons or to further the military's mission." A service member "freely agrees when joining the military to relinquish autonomy" to the interests of the unit or mission. Furthermore, they are aware "as they approach actual fighting, [that] their autonomy dramatically decreases." Thus, when individuals join the military "they implicitly agree to subordinate their own interests and autonomy to the military when necessary for the unit or the mission." In response, the military promises "to protect them to the degree possible during combat."
In this context, the argument against allowing service members to grant or withhold informed consent becomes clear.
If the military gave servicepersons the choice of accepting or refusing to take such compounds, those who chose not to take the compounds would violate the overriding obligation to the unit and the military, and the military would violate its obligation to them.Those who refused could either leave the combat situation or remain without protection. The undesirable consequences would be to increase the danger to other service personnel, to reduce the likelihood of a successful mission, and potentially to place themselves at risk.
On the assumption that the benefits of taking the investigational compounds greatly exceed the risks, as evaluated as objectively as possible by civilian reviewers outside of DoD,
permitting servicepersons to grant or withhold informed consent cannot be ethically justifiable in principle [emphasis in original] because this would give servicepersons' individual autonomy priority over their obligations to the unit and society, thus unnecessarily endangering other servicepersons and the success of the mission.Two of the most prominent critics of the Interim Rule, Annas and Grodin, of Boston University, responded to Howe and Martin in the same issue of the Hastings Center Report (Annas and Grodin, 1991). They characterized the Interim Rule as a radical departure from long-standing DoD policy by requiring U.S. troops to take unapproved drugs and vaccines without consent--"informed or otherwise." That long-standing policy began in 1953, they argue, when DoD adopted the Nuremberg Code as official policy, including the essential requirement of informed consent for research subjects.[2] DoD policy had been consistent since 1953, then, and had included the adoption in 1986 of the "Proposed Model Federal Policy for the Protection of Human Subjects," which became the Common Rule.
Annas and Grodin rejected the argument that these policies were irrelevant because the INDs were being given for treatment not research. "Until now," they argued, the military position had been that service personnel "must accept standard medical treatment (or face court martial)." However, they have no obligation to accept interventions "not generally recognized as standard procedures." They quoted a decision of the Army Judge Advocate General that
"only those [procedures] thoroughly tried and generally used by the medical professional that have definitely and finally passed the novel and experimental stages, and has been accepted as standard operations in surgery,"can be performed without consent. The issue to Annas and Grodin was clear: "it is the experimental or investigational nature of the intervention, not the intent [emphasis added] of the physician or researcher, that determines whether or not an intervention is research or therapy." Moreover, the absence of alternative treatments "does not convert an investigational intervention into a therapeutic one." Rather, consent is required for investigational drugs not only by the Nuremberg Code, but by the Declaration of Helsinki, HHS regulations, and FDA regulations.[3] Moreover, "no exception [emphasis in original] is made for the military or wartime in any of these codes and regulations."
Annas and Grodin attacked the DoD justifications for the Interim Rule. The argument that the investigational drugs used in the Gulf War were safe and effective and constituted the primary preventive treatment available was vitiated by DoD's request for an exception to use them without informed consent; if the interventions were treatments, no exemption and no consent would be required. Second, the second justification that consent was "not feasible" was basically an extension of Hippocratic paternalism from "the doctor knows best" to "the Army knows best" and represented a substantial assault on the rights of American servicemen and women. Third, service personnel do not subordinate themselves entirely to the military mission or to the welfare of their fellow soldiers. They have both a right and a duty to disobey an unlawful order. But was not the order to take investigational drugs the same as an order to wear protective clothing and equipment? "The answer is no," they responded.
The helmet or flak jacket cannot injure the serviceperson, has no side effects, and can only protect. The unproven vaccine, on the other hand, can actually cause more problems than it solves and can cause more injury to individual troops than the biological agent it is designed to protect against.They extended their argument even further: "Ground troops are not asked if they want to advance; they are ordered to advance even if many of them will likely perish. But the order to march does not justify the order to submit to experimental vaccines."
Finally, Annas and Grodin argued, in response to the DoD argument that the Nuremberg Code and the Declaration of Helsinki did not apply "to the wartime military," the Nuremberg Code was not articulated for the expedient purposes of "any country, army, or research endeavor." Rather, its principles
were derived from [quoting the Code] "the principles of the law of nations as they result from the usages established among civilized peoples, from the laws of humanity, and from the dictates of public conscience." Those who fight to protect these basic human rights should be protected by them.Also in the Hastings Center Report, Levine (1991) of Yale University School of Medicine, long-time student of institutional review boards and informed consent, thanked Howe and Martin for their "careful account of the ethical justifications" behind the interim final rule. He took up "the allegation" that DoD's planned use of investigational drugs was research. He cited the Belmont Report's definition of research as "an activity designed to test a hypothesis, permit conclusions to be drawn, and thereby to develop or contribute to generalizable knowledge" and that of DHHS regulations "as a systematic investigation designed to develop generalizable knowledge." The intended uses of INDs in the Gulf clearly did not constitute research but were intended to treat or prevent the "horrific" injuries that might be caused by chemical or biological warfare agents. These uses conformed to the definition of medical practice of the Belmont Report and also noted that the U.S. District Court had dismissed Doe v. Sullivan, in part, because of the "lack of conformity to the definition of research--within the meaning intended or specified in any relevant law." The frequent failure of many commentators to make the distinction between research and treatment stemmed, in Levine's view, from "incautious use of poorly defined terms and concepts" by many discussants. Investigational, in its denotative meaning, is applied to drugs not approved by FDA for commercial distribution; however, it connotes "images of research." Moreover, the term "investigational new drug" evolved within FDA from the legal exemption authorizing distribution across state boundaries for purposes of the conduct of research and only later came to refer to the drug or vaccine itself.
On the informed consent requirement for human use of all investigational drugs, even if used for therapy outside of a research protocol, Levine reasoned that military personnel "in certain situations do not have the same rights to self-determination as civilians; they may not refuse therapies designed to maintain their effectiveness." So when DoD determined that INDs were the best preventive or therapeutic treatment against CW/BW agents, "administering them without informed consent would have been in accord with customary military procedure." In response to critics of the Interim Rule who claimed that it was "feasible" to obtain consent in the Gulf, Levine concurred that it would be "doable" in the "very limited sense" of obtaining 500,000 signatures on consent forms, but that it would not be "reasonable" or "suitable."
In 1994, as Congress increased its postwar interest in the Gulf War, Arthur Caplan, prominent University of Pennsylvania bioethicist and later a member of the PAC on Gulf War Veterans' Illnesses, testified before a Senate committee (1994). The issue of whether the use of investigational drugs and vaccines in the Gulf for CW/BW defense constituted research or treatment was a settled issue in his view. It was research. Research, he argued, was defined by intent: "The standing definition of research in American law looks to intent to define research. If a goal of undertaking and intervention with a human being is to generate new knowledge then that activity counts as research."
Although the conduct of research on military personnel was "most difficult" when "threatened with the prospect of war or . . . actually engaged in an armed conflict," Caplan testified, the standards for such research were clear. They derived from the Nuremberg Code, which had been "set down as a direct response to experiments conducted under conditions of war," and which made
no exception [emphasis added] for research conducted in the context of war. . . .The essential requirement of informed consent, Caplan argued, "is not modulated by the existence of a state of war."The enormously important goal of protecting the nation's security is not held to be a value that is so overriding as to obliterate the individual subjects' rights. The Code states clearly and unambiguously that everyone involved in research is to be so informed and that they are to have the right to give or withhold their consent to that research.
Although some military efforts were clearly research, Caplan acknowledged that the military use in the Gulf War of PB and BT was arguably to protect troops from lethal chemical and biological agents and thus not research. For Caplan, these arguments were "not persuasive." Why? For one thing, he asserted, the use of "unapproved, unlicensed agents clearly" was understood by FDA and DoD to be research in that both agencies recognized the need to seek waivers from prevailing informed consent requirements. Moreover, it was understood by these regulatory and military officials that the practical reasons it was difficult to obtain consent "for the use of untested, unproven agents in large populations deployed in trying environments under battlefield conditions meant that the use of these agents had to be seen as experimental."
Although it could be argued that those seeking to use investigational agents to obtain prophylactic benefit for military personnel "did so by weighing carefully the cost, risk, and potential benefits," neither the conduct of such analysis, nor results of such analysis that favored the use of investigational agents, nor the intent to use such agents to "benefit, treat, or prevent harm" could "transform an experimental intervention into a therapy."
However, Caplan expressed reservation about this definition. Reliance on the regulatory definition of research as "intent to create generalizable knowledge" was fine as far as it went but it was not adequate to capture activities that are
manifestly experimental but are not conducted with the intent of generating new knowledge. . . . This is obviously what took place in the Gulf with anti-bot toxoid [sic] and pyridostigmine bromide. These agents were used in large populations for purposes other than those for which they were originally designed in circumstances under which they had never before been tried.Moreover, given uncertainty about what chemical or biological agents might have been deployed by Iraq, PB and BT "may have had no efficacy or might actually have had an adverse effect in the case of the utilization of certain nerve gas agents." Furthermore, efficacy was open to question in Caplan's view because the actual administration of these agents deviated from the normal three-shot regime (for BT). "It seems plain," he stated, that the most likely reason for the decision to grant the waiver was "that there was a chance the agents would do more good than harm but that the efficacy of these agents to prevent harm was seen as far from certain."
In the final paragraph of his testimony, Caplan offered this advice:
Those engaged in decisions to use unproven, untested, unlicensed or otherwise experimental or research agents in the context of biological and chemical warfare or other battlefield circumstances must understand that conditions of war may lead to decisions which, while undertaken with the best of motives, may not serve to protect the welfare of military personnel or permit the completion of the assigned military mission. Americans when faced with a challenge often feel that it is better to try something rather than to do nothing. The history of human experimentation in this century shows that often, when faced with a crisis, doing nothing in the face of uncertainty can be the most prudent course of action to follow.In summary, the critics make these arguments. First, they emphasize the unknowns of research. Annas and Grodin (1991), for example, state: "Research is research because we don't know the consequences, risks, and benefits of intervention." Caplan (1991) argues that the "manifestly experimental" nature of investigational drugs is clear because they "were used in large populations for purposes other than those for which they were originally designed in circumstances under which they had never before been tried."[4] Further, Caplan argues, the research nature of the use of PB and BT in the Gulf is "cemented by the obvious uncertainty that accompanied the utilization of these agents as to the efficacy that they would have in the field." Uncertainty about the consequences of nonuse in the face of enemy capabilities and intentions receives no attention.
Second, the critics attack the inadequacy of intent to differentiate between research and treatment. Here they rely on the Belmont Report as the authoritative statement that research is an activity whose purpose is to generate generalizable knowledge. Thus Caplan (1991) argues that, if intent were controlling "those who do research would merely have to change their intentions and they could succeed in making the most innovative and experimental medical interventions into therapies merely by a change of mind." In a similar vein, Annas and Grodin (1991) write: "The point is clear. It is the experimental or investigational nature of the intervention, not the intent of the physician or researcher, that determines whether or not an intervention is research or therapy." Both might have invoked Sabiston and Robertson at this juncture, neither of whom found intent especially helpful. Also overlooked was the fact that few advocates for the use of investigational drugs for CW/BW defense rely on intent as the sole criterion differentiating between research and treatment, a matter we discuss at greater length below.
Third, the critics argue that acceptance of the requirements of regulation constitutes de facto recognition that research is involved. Caplan testified: "The use of unapproved, unlicensed agents was clearly understood by FDA and DoD to be research inasmuch as both agencies recognized the need to seek waivers from prevailing informed consent requirements." Such a categorical statement denies the possibility that both parties might be proceeding on the basis that ambiguity exists regarding the definition of research and that the relation between research and investigational is an arguable policy issue, but that the parties agree that deference should be given to established regulations even while seeking to modify those regulations to accommodate challenges unanticipated when they were adopted. Martin (1996), responding to this point, said, "Referring to these drugs and vaccines as "investigational" is in accordance with Food and Drug Administration (FDA) regulations and is not a definitive statement regarding the scientific information available about the INDs." In addition, both Annas and Grodin, as well as Caplan, note that the Nuremberg Code was conceived in the wake of war, both as a reaction to the despicable war crimes of the Nazi doctors and as an effort to articulate "universal principles" governing human experimentation. Then, since the code made no exceptions for research in wartime, no exceptions should be tolerated for such situations as the Gulf War. They reject as not pertinent the fact that the Nuremberg Code was articulated with reference to a quite different empirical situation than that existing in the Gulf in 1990 and 1991.
The purpose of the critics' line of argument is clear: If investigational means research under any and all circumstances, informed consent is required at all times. Consequently, a willingness to acknowledge that there may be distinctions among various drugs FDA has classified as "investigational" is ruled out a priori. In short, the experiment-therapy continuum does not represent a gray zone for the critics, but investigational lies at all times on the research side of a bright orange line.
If the weight of the investigational designation falls heavily on requiring informed consent, how did DoD define both PB and BT as treatment, not research, and justify waiving informed consent? The arguments, which were laid out in the Mendez letter of October 30, 1990, were as follows. First, DoD highlighted the Iraqi threat and the need to prepare in advance for the uncertainty of the enemy's capabilities and intentions. This was especially true for Saddam Hussein, who had previously used CW/BW agents in Iraq's war with Iran as well as against ethnic groups in Iraq itself. Second, there was the ethical commitment to protect U.S. military personnel from the threat that was at the heart of seeking approval to use "the best preventive or therapeutic treatment" available. Third, in scientific and clinical terms, the drugs under consideration, although classified by FDA as investigational, were not "exotic new drugs"; they were not novel. Rather, they had "well-established uses," both preventive and therapeutic. The DoD medical establishment had concluded that these drugs were the best available way to protect the troops. The safety of these drugs had been established by prior use, human data supported the effectiveness of BT and animal data provided a defensible scientific basis for the effectiveness of PB. Consequently, DoD was not proposing to engage in clinical investigation but to use these drugs as the only medical therapies available for the treatment (or pretreatment) of potentially lethal CW/BW agents. Hundreds of thousands of troops were at risk in the Gulf and the issue for DoD was how it should be fulfill its obligation to protect them, "How do we take care of people?"
In DoD's judgment, a gray zone existed in which products legally classified as investigational could be justifiably represented as having nonresearch, therapeutic uses. That gray zone was legally provided for in Section 505(i) of the FDCA. This section requires that investigators using drugs for investigational purposes
inform any human beings to whom such drugs . . . are being administered, or their representatives, that such drugs are being used for investigational purposes and will obtain the consent of such human beings or their representatives, except where they deem it not feasible or, in their professional judgment, contrary to the best interests of such human beings [emphasis added].This statutory provision for exceptions from the requirements of informed consent provides the basis for a "not feasible" determination and for DoD requesting waiver of informed consent of investigational drugs in the Gulf War.
Most important, FDA places substantial weight on methodology as a defining feature of clinical investigations. Section 505(d) of the FDCA states that the secretary "shall refuse to approve" an [NDA] application if safety and effectiveness standards are not met. The criterion of "safe for use" is that a drug be supported by "adequate tests by all methods reasonably applicable"; the effectiveness criterion is that "substantial evidence that the drug will have the effect it purports or is represented to have" is met. Substantial evidence is further defined as
evidence consisting of adequate and well-controlled studies, including clinical investigations, by experts qualified by scientific training and experience to evaluate the effectiveness of the drug involved, on the basis of which it could fairly and responsibly be concluded by such experts that the drug will have the effect it purports or is represented to have under the conditions of use prescribed, recommended, or suggested in the labeling or proposed labeling thereof.[5]Thus, the FDCA, augmented by FDA regulations and guidelines, approaches the definition of clinical investigations not only with respect to intent but also with reference to statistical and clinical methodology and to the qualifications of investigators (which include both training and experience). This approach, constrained by the law, is far richer and more empirically grounded than that of the useful, but limited, Belmont Report.
[1]Howe was associate professor of psychiatry and director of programs in medical ethics, Department of Psychiatry, Uniformed Services University of the Health Sciences. Martin, a physician and career Public Health Service officer, was then Deputy Assistant Secretary of Defense for Professional Affairs and Quality Assurance, Health Affairs.
[2]That policy, however, was classified as Top Secret until 1975, when then-U.S. Army Surgeon General Richard R. Taylor testified before a subcommittee of the U.S. Senate Judiciary Committee dealing with military experiments:
The basic Department of Defense policy governing medical experiments was promulgated by the Secretary of Defense on 26 February 1953. This policy is based on the Nuremberg Code of 1947, which followed the war crimes trials. . . . [A]n individual participating as a subject is required to . . . give voluntary written informed consent without coercion. . . . These basic moral, ethical, and legal principles . . . are common to the regulations of the military departments.[3]Annas and Grodin were either unaware that FDA regulations allowed exceptions or chose to ignore this fact.
[4]This view, taken to its logical conclusion, would classify early use of an FDA-approved drug as "experimental" because the small sample sizes in Phase I, II, and III clinical trials were inadequate to reveal rare side effects or long-term effectiveness in the intended population of normal users.
[5]The importance of methodology, and of the quality of evidence as judged by experts qualified by training and experience to make scientific determinations, is sufficiently great that FDA's CDER has a 125-page Guideline for the Format and Content of the Clinical and Statistical Sections of New Drug Applications, published in 1988, which provides detailed instructions about reporting results of clinical investigations. In addition, this "guideline" requires that each NDA list "all investigators supplied with the drug substance or drug product by the applicant or known to have investigated the drug" and indicate the kinds of studies each investigator has conducted.